
MIOPATIAS MITOCONDRIAIS
As miopatias mitocondriais são distúrbios musculares herdados através de genes defeituosos nas mitocôndrias ou nos genes nucleares que controlam o funcionamento das mitocôndrias. Os músculos e órgãos, como o cérebro, nervos e retinas, são afetados.
Síndrome de Kearns-Sayre (SKS) e Oftalmoplegia Externa Progressiva Crônica (OEPC)
Ptose e oftalmoparesia são aspectos centrais da SKS e da OEPC. A OEPC é uma miopatia pura com
fraqueza extra-ocular que se manifesta por ptose e oftalmoparesia. Outros músculos podem estar afetados,
causando fraqueza da face, orofaringe e membros. Ao contrário, a SKS é uma doença de múltiplos sistemas, de
início na juventude ou idade adulta, que produz oftalmoparesia, degeneração pigmentar da retina, e
combinações variáveis de bloqueio de condução cardíaca e hiperproteinorraquia (> 100 mg/dL), além de ataxia
cerebelar. Esses pacientes podem ainda ter baixa estatura e diabete melito, hipoparatiroidismo, miocárdio e
nefropatia. A biópsia muscular identifica fibras vermelhas rajadas e o defeito molecular.
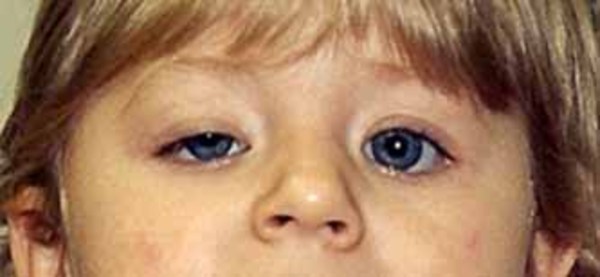
figura 1 : ptose palpebral ( palpebras caidas)
Credit: Elements of Morphology, National Human Genome Research Institute
Síndrome MELAS
A síndrome MELAS caracteriza-se por episódios de isquemia cortical na idade jovem (< 40 anos),
encefalopatia (crises epilépticas, demência ou ambas) e disfunção mitocondrial, caracterizada por acidose
láctica, fibras vermelhas rajadas ou ambas. A prevalência da mutação mais comum é de 1,4 a 16,3 casos por
100.000 pessoas. Chama a atenção o fato de o desenvolvimento infantil ser normal e haver cefaléia recorrente,
ou vômitos cíclicos. É possível o achado de fraqueza muscular miopática, intolerância ao exercício, mioclonias,
ataxia, baixa estatura e perda auditiva. É raro que mais de um membro da família tenha a síndrome completa. A
maioria é oligo ou assintomática. O padrão de imagem dos eventos isquêmicos é típico: lesões corticais que não
respeitam território vascular. Sempre que um paciente com MELAS apresenta um episódio de acidente vascular
cerebral (AVC), o diagnóstico diferencial inclui outras causas de AVC em jovem: doença das artérias carótidas
ou vertebrais, anemia falciforme, vasculopatias, distúrbios das lipoproteínas, câncer, trombose venosa, doença
de Moyamoya, enxaqueca complicada e homocistinúria.
Síndrome MERRF
A ocorrência de epilepsia, mioclonias, ataxia e biópsia muscular mostrando fibras vermelhas rajadas
define o diagnóstico de MERRF. Também pode ocorrer perda auditiva, demência, neuropatia periférica, baixa
estatura, intolerância ao exercício, lipomas e acidose láctica. O diagnóstico é confirmado pela pesquisa da
mutação genética. O diagnóstico diferencial inclui outras causas de epilepsias mioclônicas progressivas, como
síndrome de Unverricht-Lundborg, doença dos corpos de Lafora, lipofuscinose ceróide neuronal e sialidose.
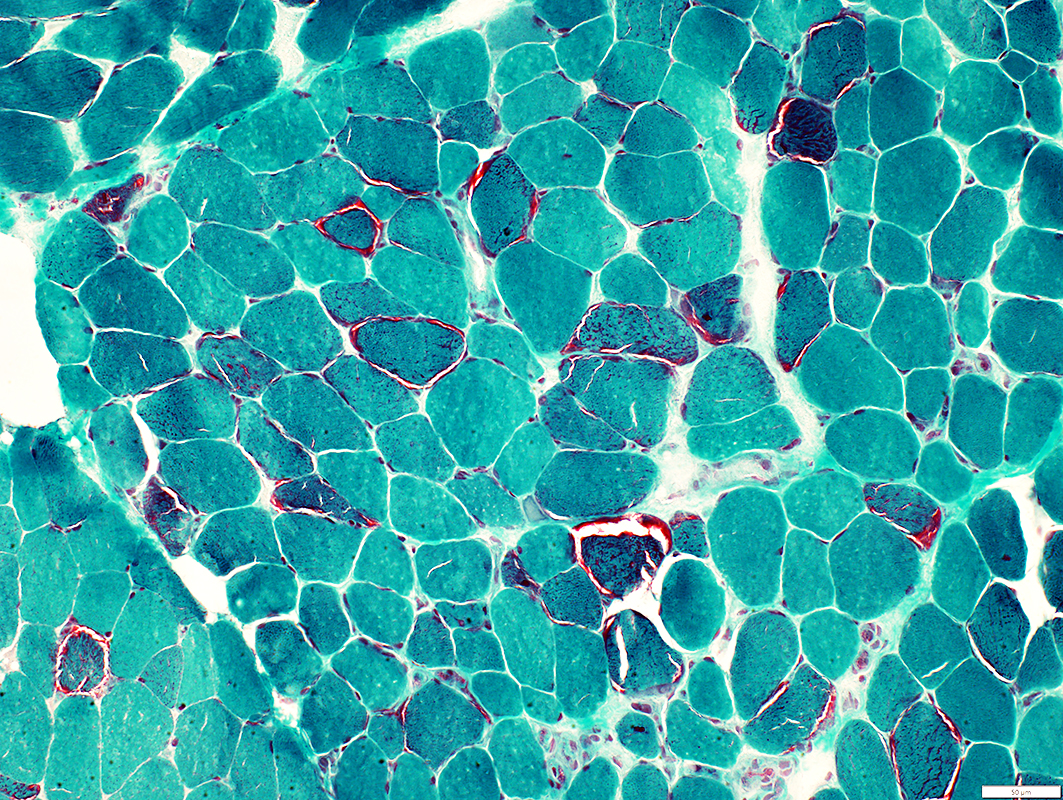 figura 2 : fibras vermelhas rasgadas (red ragged fiber) caracterista da biopsia muscular nas mitocondriopatias
figura 2 : fibras vermelhas rasgadas (red ragged fiber) caracterista da biopsia muscular nas mitocondriopatias
credito https://neuromuscular.wustl.edu/pathol/melas.htm.
Síndrome de Leigh de Herança Materna
Pacientes com mutação maior que 90% da subunidade 6 do complexo V da cadeia respiratória
desenvolverão um quadro devastador de encefalomiopatia, caracterizada por regressão do desenvolvimento
psicomotor, crises convulsivas, acidose láctica e lesões necrotizantes subagudas nos gânglios da base e outras
estruturas da substância cinzenta, na linha média do cérebro e tronco. Isso caracteriza a síndrome de Leigh de
herança materna (MILS). A ressonância magnética mostra lesões características da região periaquedutal do
mesencéfalo e ponte, e no bulbo, adjacente ao quarto ventrículo. Podem afetar outras estruturas do sistema
nervoso central e periférico. O diagnóstico diferencial inclui a doença de Refsum, a abetalipoproteinemia e
outras doenças mitocondriais.
Referencias Bibliograficas
Schon EA, DiMauro S. Medicinal and genetic approaches to the treatment of mitochondrial disease. Curr
Med Chem 2003;10:2523-33.
United Mitochodnrial Disease Foundation. Types of Mitochondrial Diseases. Available
from: http://www.umdf.org/types/
Greaves LC, Taylor RW. Mitochondrial DNA mutations in human disease. IUBMB Life 2006;58:143-
51.
Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders--
past, present and future. Biochim Biophys Acta 2004;1659:115-20.
Saneto RP, Sedensky MM. Mitochondrial disease in childhood: mtDNA encoded. Neurotherapeutics
2013;10:199-211.
Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull 2013;106:135-59.